

This document discusses how to deal with one or more MUMmer alignments failing. The corresponding MUMmer documentation is v3 and v4.
MUMmer files
When/data/seq_results/<project-name1>-to-<project-name2>/alignFor example,
data/seq_results/demo_seq_to_demo_seq2/align> ls all.done demo_seq_cc.demo_seq2_f2.mum demo_seq_cc.demo_seq2_f1.mum demo_seq_cc.demo_seq2_f2.mum.done demo_seq_cc.demo_seq2_f1.mum.done
All MUMmer files but the ".mum" are removed by symap; if you prefer them not to be removed, start symap with the "-mum" command line parameter, i.e. ./symap -mum
The log files are in the /logs/<project-name1>-to-<project-name2> directory.
Finding the problem
| Using MUMmer4 | Executables | Go to top |
If the MUMmer alignment fails, first check the Executables. If they are okay, then inspect the log files. The log files are as follows:
symap_5/
error.log # a SyMAP error will write its trace data into this file and list failed MUMmer
logs/
<p1>-to-<p2>/ # one directory per project-to-project alignment
<p1_cc.p2_f1>.log # MUMmer terminal output - one file per MUMmer process
<p1_cc.p2_f2>.log # fn is n=1,2... for number of processes, e.g f2 is 2nd process
symap.log # keeps most of the SyMAP output shown on the terminal for this A&S
e.g.
demo_seq_to_demo_seq2/
demo_seq_cc.demo_seq2_f1.log # MUMmer output directed to this file
demo_seq_cc.demo_seq2_f2.log # same for the second mummer execution
symap.log # SyMAP terminal output
→ If an alignment is listed as failed in the error.log file,
the corresponding <p1_cc.p2_fn>.log file will contain the MUMmer error
(see example mummer log).
→
If the error is not found in the log files or it is not clear, try the following:
Copy the command from the terminal (or log file), and paste it on a new terminal line to execute, e.g.
(the following is one line wrapped around):
ext/mummer/mac/promer -p data/seq_results/demo_seq_to_demo_seq2/align/demo_seq_cc.demo_seq2_f2.promer data/seq_results/demo_seq_to_demo_seq2/tmp/demo_seq2/demo_seq2_f2.fa data/seq_results/demo_seq_to_demo_seq2/tmp/demo_seq/demo_seq_cc.faThis shows promer output directly on the terminal. Typically the problem is not enough memory.
Using MUMmer4
Sometimes when MUMmer v3 fails, MUMmer v4 will work. MUMmer4 is included in SyMAP package with a fix to promer. Enter the ext/mummer4 directory and follow the instructions in the README.Executables
The alignment programs are provided in the symap/ext directory. There are executables for 64-bit Linux and 64-bit MacOS. SyMAP will select the correct directory for the machine you are running from, i.e. you do not need to do anything. See System Guide for details.When SyMAP creates a database, it (1) checks the MySQL variables, and (2) checks that the external programs are executable. If you see a message like:
***Error - file is not executable: ext/mummer/mac/promerExecute:
> chmod 755 ext/mummer/mac/promerExecute the program from the command line to make sure it works on you machine, e.g.
>./ext/mummer/mac/promer
USAGE: promer [options] <Reference> <Query>
Try './ext/mummer/mac/promer -h' for more information.
The above shows that the promer code will execute on my MacOS.
Executable on MacOS: Any executable that has not been okayed by Apple results in the error message
cannot be opened because the developer cannot be verifiedSee MacOS External for the fix.
Out of memory
| Limit CPUs and uncheck Concat | Not-masked or Soft-masked | One or more alignments fail | Go to top |
A MUMmer failure is typically from insufficient memory.
If an alignment fails immediately, and if the last line of the first <alignment>.log file is:
1: PREPARING DATAthe reason is probably that your machine does not have near enough memory as MUMmer could not even prepare the data.
Additionally, the following errors typically indicates a memory problem:
Alignment program error code: 141 20220512|075853|6007| ERROR: mummer and/or mgaps returned non-zero, please file a bug reportor
#..........................ERROR: mummer and/or mgaps returned non-zero, please file a bug report Alignment program error code: 1The error code will appear on the terminal and the MUMmer log file, but not in symap/logs/<..>/symap.log.
Limit CPUs and uncheck Concat
There is no straight-forward way to know if you have enough memory as it depends on the size and complexity of the two genomes being compared. If you think memory may be tight or MUMmer produced an error as shown in the section above, first try running again with reduced CPUs and unchecked- On the
Project Manager panel, limit the number of CPUs, as each CPU uses a considerable amount of memory (e.g. 4 CPUs could collectively use 20GB of memory at once). -
In the
Project Parameters panel, uncheckConcat to reduce the size of the input files to MUMmer. See Concat for a description of this option.
Not-masked or Soft-masked
A memory problem can occur if the genome sequence is not masked or only soft-masked. Either: (1) change the sequence to hard-masked, or (2) set the Pair Parameters parameterOne or more fails
Sometimes just one or a few of the alignment processes will fail. You will see a line such as:Error: Running command: /Users/cari/Workspace/symap_5/ext/mummer/mac/promer -p data/seq_results/demo_seq_to_demo_seq2/align/demo_seq_cc.demo_seq2_f2.promer data/seq_results/demo_seq_to_demo_seq2/tmp/demo_seq2/demo_seq2_f2.fa data/seq_results/demo_seq_to_demo_seq2/tmp/demo_seq/demo_seq_cc.fa
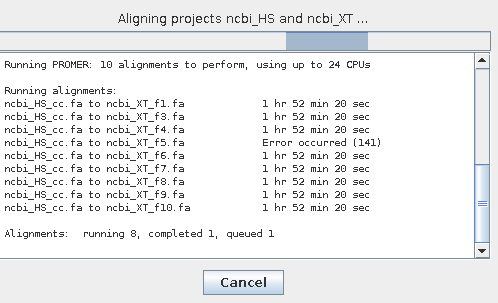
|
You will see the failure on the dialog box as is
shown on the left. The remaining processes will continue. When all processes are complete,
you will see a "?" for the pair as shown on the below.
Select the "?" followed by
|

{kind=link}
Running MUMmer from the command line
| Example | Go to top |
- If your chromosomes are large, split the sequence file into chromosome files using xToSymap.
Process each chromosome file for the first genome with each chromosome file for the second genome.
- Create a directory data/seq_results/<proj1-to-proj2>/align where proj1 is
alphabetically less than proj2.
- Running MUMmer: The query must be alphanumerically less than reference. For the promer command:
USAGE: promer [options] <Reference> <Query> e.g. promer proj2 proj1
See the commands for the Example below. Replace the demo names with your project/chromosome names. If you have many chromosome pairs to process, put them in a script, e.g. demo commands.
- Promer: The output of promer is input to "show-coords -dlkT".
- Nucmer: The output of nucmer is input to "show-coords -dlT".
To view the MUMmer parameters, see MUMmer parameters.
- Result files:
- The result files must have suffix ".mum"
- The ".mum" files must be in the directory data/seq_results/<proj1-to-proj2>/align.
- In the <proj1-to-proj2>/align directory, execute:
touch all.done
This creates a file, which indicates to SyMAP that the alignments are done and to process the files in the directory ending with ".mum".
- The result files must have suffix ".mum"
- When you run
Selected Pair , SyMAP will recognize the files and use them to build the synteny blocks.
Example
This example will use MUMmer for the loaded projects demo_seq and demo_seq2. First, remove the directory data/seq_results/demo_seq_to_demo_seq2.Demo_seq has chr3 and chr5 in the file genomic.a and demo_seq2 has files chr1.seq.gz and chr3.seq. The commands are as follows:
gunzip data/seq/demo_seq2/sequence/chr1.seq.gz # MUMmer does not process zipped files
cd data/seq_results
mkdir demo_seq_to_demo_seq2
mkdir demo_seq_to_demo_seq2/align
touch demo_seq_to_demo_seq2/align/all.done
cd ../..
ext/mummer/mac/promer -p data/seq_results/demo_seq_to_demo_seq2/align/results.promer
data/seq/demo_seq2/sequence/chr1.seq data/seq/demo_seq/sequence/genomic.fa
ext/mummer/mac/show-coords -dlkTH data/seq_results/demo_seq_to_demo_seq2/align/results.promer.delta
>data/seq_results/demo_seq_to_demo_seq2/align/seq1chr1.mum
See script for the full set of mummer command to process all sequence data.
When symap is started and demo_seq and demo_seq2 selected, there will be a "A" in their cell; select it followed by
This demo has been fully tested with
Getting Help
If none of these suggestions fix your problem, email cas1@arizona.edu with the following files (described in MUMmer files):- error.log
- logs/<p1>-to-<p2>/symap.log
- logs/<p1>-to-<p2>/<p1_cc.p2.fn>.log where n is the process number
- Any output to the terminal (either copy and paste into the email, or send a screen capture)
symap/error.log symap/logs/demo_seq_to_demo_seq2/symap.log symap/logs/demo_seq_to_demo_seq2/demo_seq_cc.demo_seq2_f2.log
| Go to top |
Email Comments To: cas1@arizona.edu